ggplot(data = iris, aes(x = Sepal.Length, y = Sepal.Width, colour = Species)) +
geom_point() +
labs(x = "Sepal Length (mm)", y = "Sepal Width (mm)") +
theme_light() +
theme(legend.position = "bottom")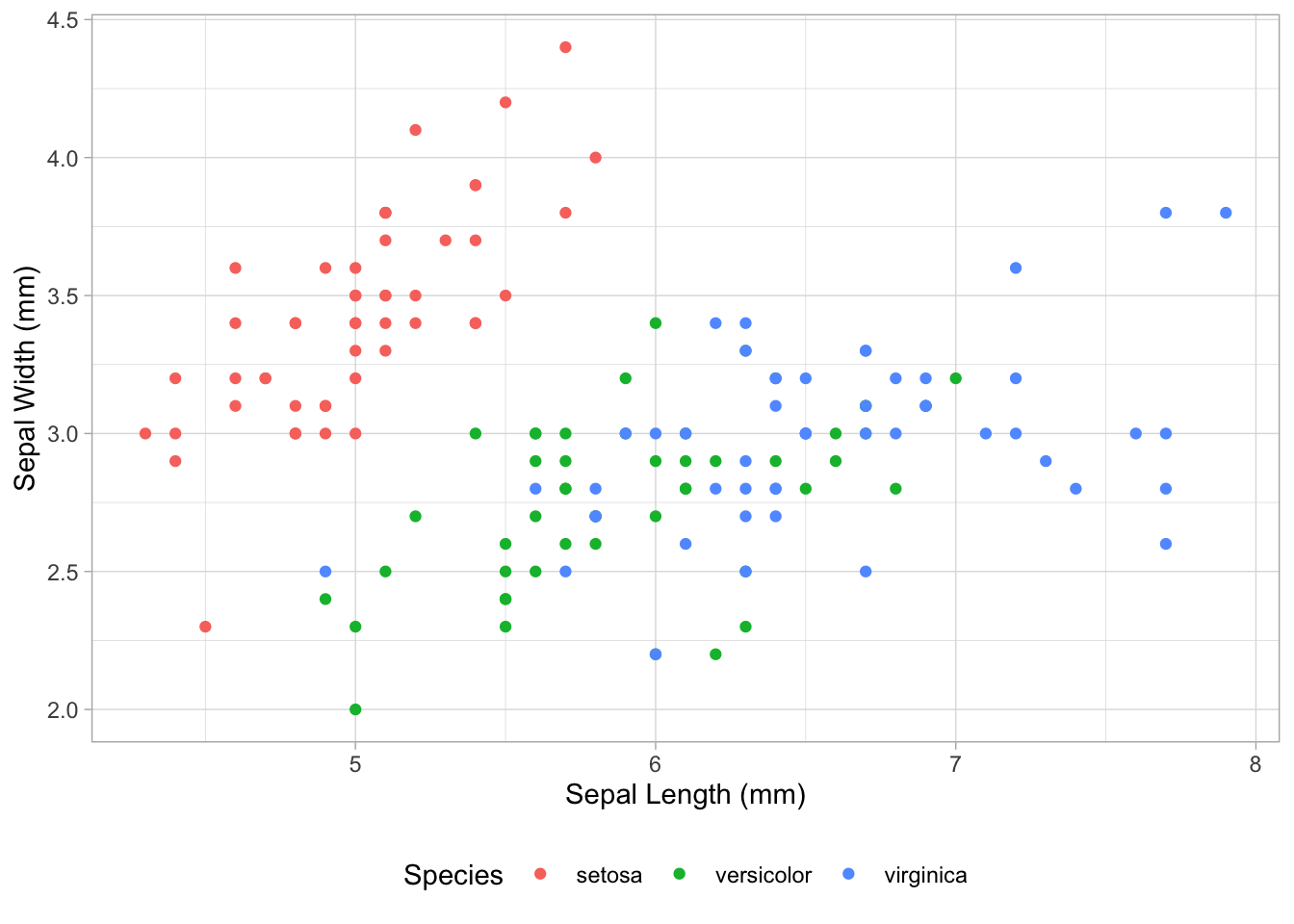
irisSolution
____.Important : Exécutez ce chunk R avant de commencer pour charger les bons paquets et les données :
1 point
Dans le TP, nous utilisions souvent les données penguins. Les données elles-mêmes (c’est-à-dire penguins) sont-elles un objet R ou une fonction R ?
penguins est un objet.
1 point
Nous avons utilisé mean() pour calculer la moyenne, par exemple d’une colonne. Est-ce que mean() est une fonction R ou un objet R ?
mean() est une fonction.
2 points
Les données iris sont aussi souvent utilisées dans les exemples. Elles contiennent des données pour différentes espèces d’iris, par exemple des mesures de la largeur et de la longueur du pétale et du sépale :

Créer un graphique avec ggplot() : à partir des données iris, représentez la colonne Sepal.Length sur l’axe x et Sepal.Width sur l’axe y. Utilisez des couleurs différentes pour les espèces Species. Vous pouvez consulter les données dans l’onglet environnement. Remplacez uniquement les ____ !
ggplot(data = iris, aes(x = Sepal.Length, y = Sepal.Width, colour = Species)) +
geom_point() +
labs(x = "Sepal Length (mm)", y = "Sepal Width (mm)") +
theme_light() +
theme(legend.position = "bottom")
4 points
2 points
Regardez le graphique que vous venez de réaliser. Si vous n’avez pas réussi à le faire, demandez-moi et je vous le montrerai. Pensez-vous qu’il existe une relation entre la longueur et la largeur des sépales ? Décrivez le type de relation (positive, négative, pas de relation).
Il existe une relation positive entre la longueur des sépales (Sepal.Length) et leur largeur (Sepal.Width).
2 points
Pensez-vous qu’il est biologiquement et statistiquement raisonnable d’inclure l’espèce comme variable explicative dans un modèle ? Par exemple si vous vous attendez à une pente différente selon l’espèce.
Il semble biologiquement plausible qu’il existe des différences dans la forme des sépales (définie par leur longueur et leur largeur) entre les espèces. Il est donc logique d’inclure la variable Species dans un modèle.
D’après le graphique, la relation entre Sepal.Length et Sepal.Width semble dépendre de l’Species. Il est donc statistiquement pertinent de prendre en compte l’Species dans le modèle de régression, soit pour inclure des pentes différentes entre les espèces, soit pour considérer des ordonnées à l’origine différentes.
1 point
Calculez pour chaque espèce (nom de colonne Species) la moyenne, le minimum et le maximum de la longueur du sépale (nom de colonne Sepal.Length) des données iris. Remplacez uniquement les ____ :
iris %>%
group_by(Species) %>%
summarise(
mean_sepal_length = mean(Sepal.Length),
min_sepal_length = min(Sepal.Length),
max_sepal_length = max(Sepal.Length)
)# A tibble: 3 × 4
Species mean_sepal_length min_sepal_length max_sepal_length
<fct> <dbl> <dbl> <dbl>
1 setosa 5.01 4.3 5.8
2 versicolor 5.94 4.9 7
3 virginica 6.59 4.9 7.96 points
Considérez le modèle et la sortie suivants :
# model
m_iris <- lm(Sepal.Width ~ Species, data = iris)
# anova output
anova(m_iris)Analysis of Variance Table
Response: Sepal.Width
Df Sum Sq Mean Sq F value Pr(>F)
Species 2 11.345 5.6725 49.16 < 2.2e-16 ***
Residuals 147 16.962 0.1154
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 11 point
Quelle est la variable réponse (la variable que vous voulez prédire) ?
La variable réponse est Sepal.Width.
1 point
Quelle est la variable explicative (celle que vous utilisez pour prédire la variable réponse) ?
La variable explicative est Species.
1 point
Regardez la sortie de anova() ci-dessus. La largeur des sépales diffère-t-elle statistiquement entre les espèces et pourquoi ?
Oui, la largeur des sépales varie statistiquement d’une espèce à l’autre, car la p-value (2,2e-16) est inférieure au niveau d’alpha de 0,05.
2 points
Faites un test post-hoc :
estimate_contrasts(model = m_iris, p_adjust = "tukey")We selected `contrast=c("Species")`.Marginal Contrasts Analysis
Level1 | Level2 | Difference | SE | 95% CI | t(147) | p
------------------------------------------------------------------------------
versicolor | setosa | -0.66 | 0.07 | [-0.79, -0.52] | -9.69 | < .001
virginica | setosa | -0.45 | 0.07 | [-0.59, -0.32] | -6.68 | < .001
virginica | versicolor | 0.20 | 0.07 | [ 0.07, 0.34] | 3.00 | 0.009
Variable predicted: Sepal.Width
Predictors contrasted: Species
p-value adjustment method: Tukeyet tracez les moyennes et intervalles de confiance estimés par le modèle :
estimate_means(model = m_iris) %>%
plot()We selected `by=c("Species")`.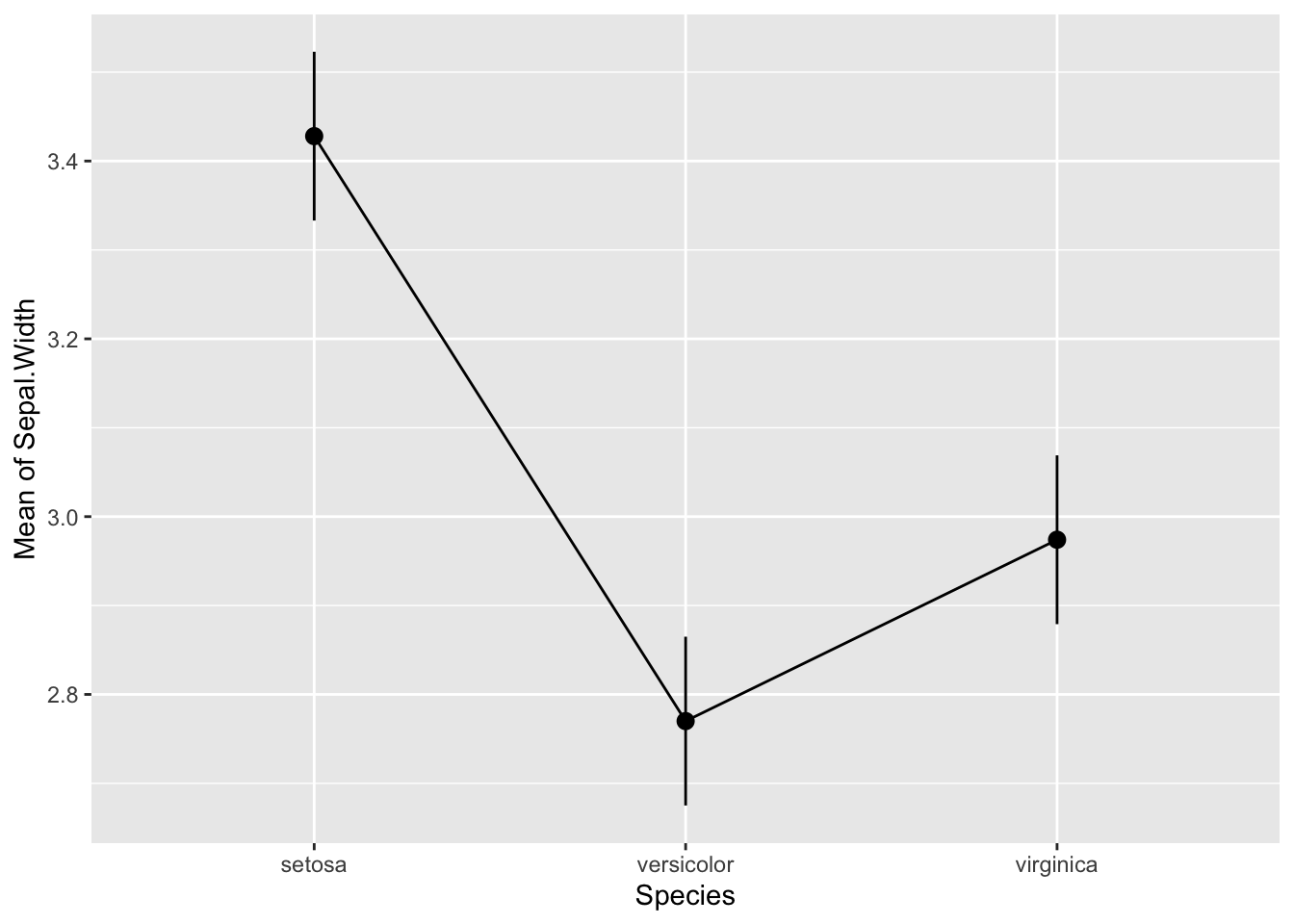
Quelles paires diffèrent significativement entre elles ?
Toutes les paires présentent des différences statistiquement significatives les unes par rapport aux autres. La différence absolue de largeur des sépales est la plus élevée entre versicolor et setosa (-0,66 mm), suivie de celle entre virginica et setosa (-0,45 mm). La différence entre virginica et versicolor est plus faible (0,20 mm) et il existe un léger chevauchement entre leurs intervalles de confiance, mais la p-value du test post-hoc (0,009) reste inférieure au niveau alpha de 0,05.
1 point
Quand on effectue de nombreux tests sur les mêmes données, comme ces tests post-hoc, pourquoi faut-il ajuster les p-values ?
Lorsqu’on effectue de nombreux tests simultanément, la probabilité de commettre au moins une erreur de type I (faux positif) augmente. C’est pourquoi il est nécessaire d’ajuster les p-values (par exemple avec la correction de Tukey) pour maintenir un taux d’erreur global acceptable.
5 points
Considérez maintenant ce modèle pour une seule espèce :
# filter data
iris_setosa <- iris %>%
filter(Species == "setosa")
# model
m_iris_setosa <- lm(Sepal.Width ~ Sepal.Length, data = iris_setosa)
# output
summary(m_iris_setosa)
Call:
lm(formula = Sepal.Width ~ Sepal.Length, data = iris_setosa)
Residuals:
Min 1Q Median 3Q Max
-0.72394 -0.18273 -0.00306 0.15738 0.51709
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -0.5694 0.5217 -1.091 0.281
Sepal.Length 0.7985 0.1040 7.681 6.71e-10 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.2565 on 48 degrees of freedom
Multiple R-squared: 0.5514, Adjusted R-squared: 0.542
F-statistic: 58.99 on 1 and 48 DF, p-value: 6.71e-101 point
La relation entre Sepal.Width et Sepal.Length est-elle positive ou négative ?
La relation entre Sepal.Width et Sepal.Length est positive, puisque la pente (0,7985) est positive.
1 point
Cette relation est-elle statistiquement significative, et pourquoi ?
Cette relation est significative, car la p-value (6,71e-10) est inférieure au niveau d’alpha de 0,05.
2 points
Quelle proportion de la variance est expliquée par le modèle (dans ce cas, par Sepal.Length) ? Donnez la valeur en pourcentage.
La valeur R² correspond à la variance expliquée par le modèle. Ici, elle est de 0,5514, ce qui correspond à 55,14 %.
1 point
Considérez l’ordonnée à l’origine (intercept) estimé et la pente donnés dans la sortie summary(). Quelle est la largeur de sépale attendue pour une longueur de sépale de 5 mm ? Vous pouvez arrondir à deux chiffres. Rappel :
\[y = a + b \cdot x\]
où \(y\) est la valeur attendue de la variable réponse, \(a\) l’ordonnée à l’origine (intercept), \(b\) la pente, et \(x\) une valeur de la variable explicative numérique.
Rappelez-vous que vous pouvez utiliser R pour ce calcul, par exemple :
3 + 5 * 7[1] 38L’ordonnée à l’origine est de -0,5694, et la pente de 0,7985:
\[ \begin{aligned} y &= -0.5694 + 0.7985 * x \\ y &= -0.5694 + 0.7985 * 5~\text{mm} \\ y &= 3.4231~\text{mm} \end{aligned} \]
4 points
1 point
Quelles sont les trois principales hypothèses des modèles linéaires ?
3 points
Expliquez brièvement chaque hypothèse.
L’indépendance des observations : Chaque observation doit être indépendante des autres — il ne doit pas y avoir de structure (par ex. mesures répétées, corrélation spatiale ou temporelle, échantillonnage hiérarchique).
L’homoscédasticité : La variance des résidus doit être constante quelle que soit la valeur de la variable explicative (variance homogène).
La normalité des résidus : Les résidus (différences entre valeurs observées et prédites) doivent suivre approximativement une distribution normale.
2 points
Regardez ce QQ-plot.
check_normality(m_iris_setosa) %>%
plot()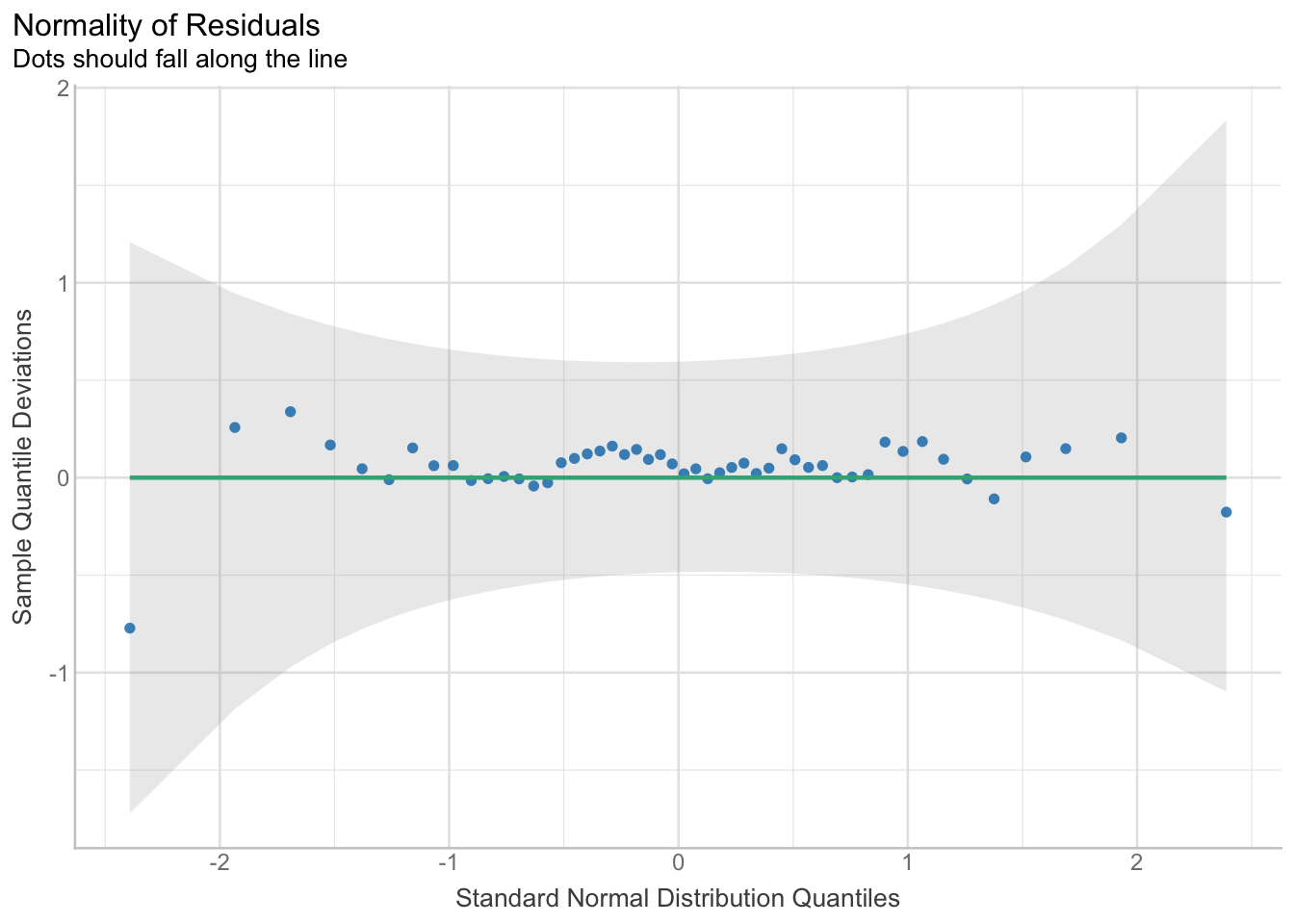
Pensez-vous que les résidus sont normalement distribués ?
Les résidus observés (représentés par des points) suivent la droite correspondant à une distribution normale des résidus. On constate de légers écarts aux extrémités, mais l’intervalle de confiance englobe cette droite, ce qui suggère que les résidus sont normalement distribués.
2 points
Regardez ce graphique qui fait partie de check_model().
# load data
dat_model_checks <- read.csv(here("exam/CC1/data/dat_model_checks.csv"))
# model
m_var <- lm(y_var ~ x_var, data = dat_model_checks)
# assess variance
check_model(m_var, check = "homogeneity")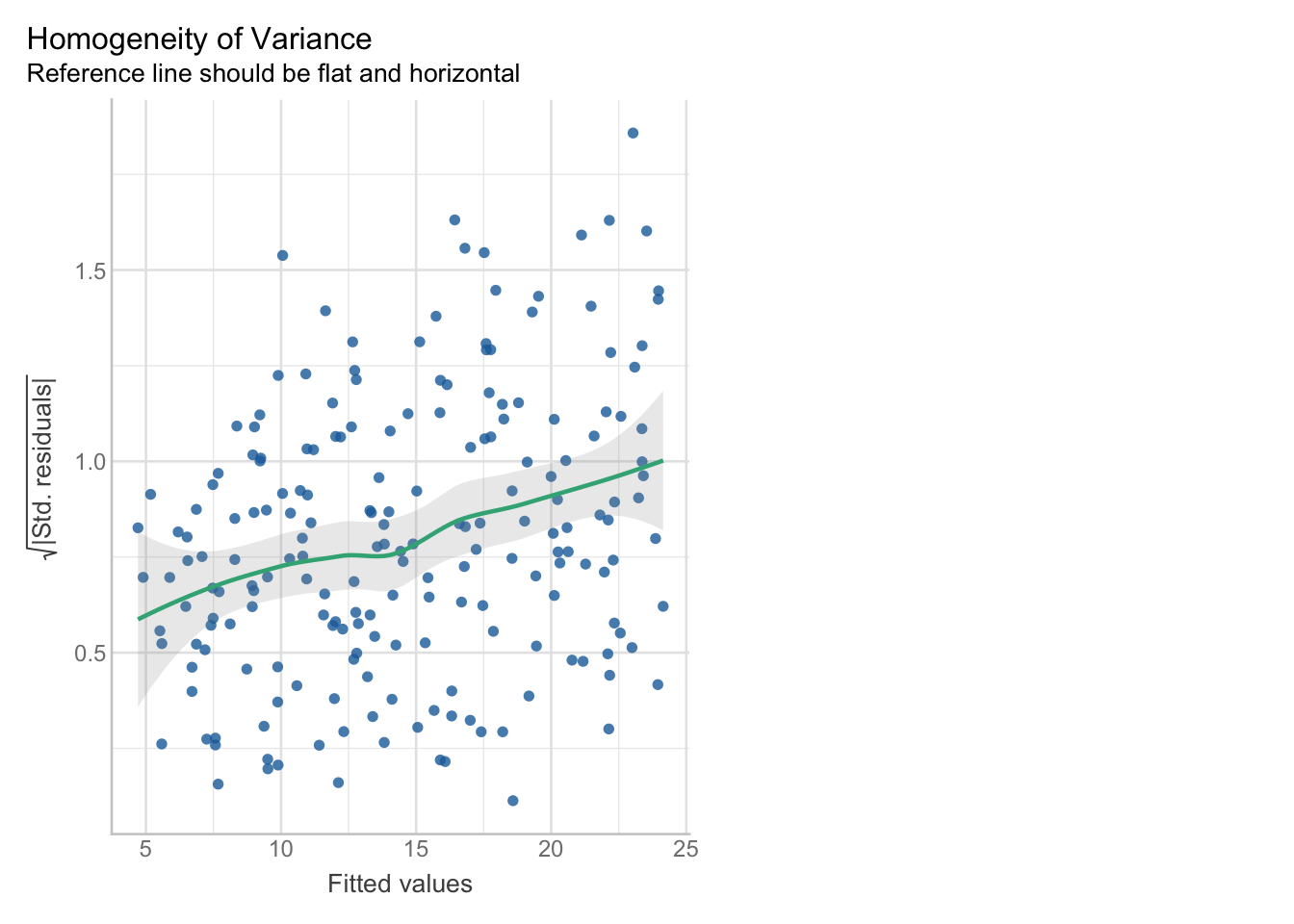
La variance est-elle égale/constante sur tout l’intervalle de la variable explicative (1 point) ?
La variance augmente avec les valeurs moyennes. On le voit à la dispersion plus importante des points pour les valeurs estimées plus élevées, ainsi qu’à la pente positive de la ligne verte. On parle alors d’hétéroscédasticité et l’hypothèse d’homoscédasticité n’est donc pas vérifiée.
Pourquoi cela impacterait-il un modèle linéaire (1 point) ?
Les modèles linéaires supposent une variance constante ; le non-respect de l’hypothèse d’homoscédasticité conduit à des estimations biaisées des p-values et des intervalles de confiance. Il est donc impossible de se fier aux résultats d’un tel modèle.
2 points
Regardez ce plan d’échantillonnage.
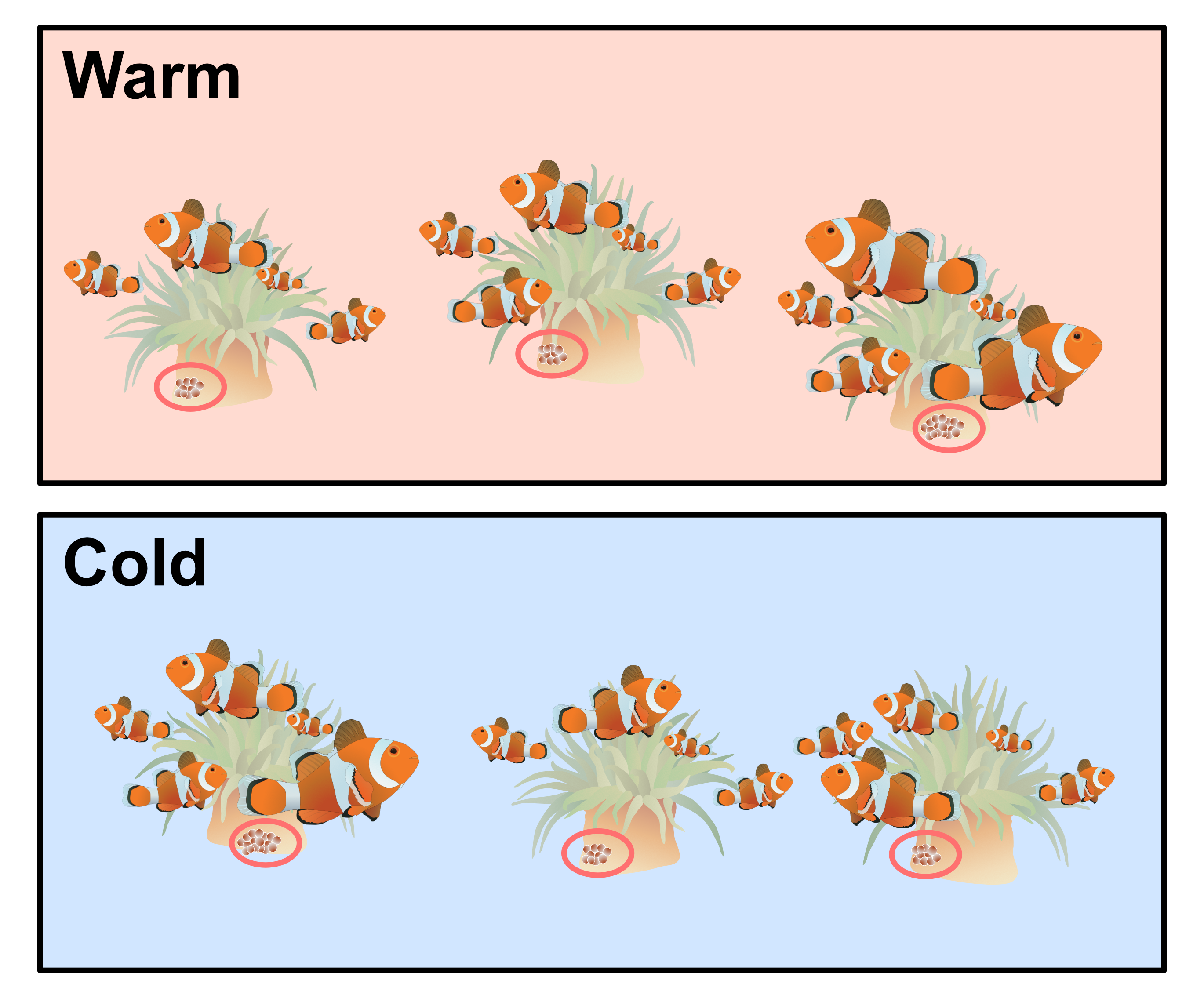
Le diamètre des oeufs de poisson-clown (dans le cercle orange à la base de l’anémone) a été mesuré sur trois anémones à deux sites avec des températures d’eau différentes (chaud en haut, froid en bas).
1 point
Pensez-vous que toutes les mesures (observations individuelles des oeufs) sont indépendantes ou y a-t-il une structure ?
Les données présentent une certaine structure. Dans le modèle, vous comparez les deux sites (vous souhaitez donc disposer de conditions différentes). Mais sur chaque site, vous avez mesuré plusieurs œufs sur trois anémones. Il s’agit de pseudo-répétitions, car elles correspondent aux conditions locales de chaque anémone, et non aux conditions générales du site !
1 point
Pourquoi est-ce important pour les modèles linéaires ?
Si le modèle considère la pseudo-réplication (réplicats au niveau de chaque anémone) comme un véritable réplicat, les p-values et les intervalles de confiance seront biaisés.
4 points en plus
1 point en plus
Prenez les données iris et évaluez l’impact de Petal.Width et Species sur Petal.Length avec un modèle linéaire incluant une interaction. Cela correspond au graphique de la Question 3. Vous devez écrire votre propre code R.
m_iris_spec <- lm(Petal.Length ~ Petal.Width * Species, data = iris)1 point en plus
Interprétez le modèle (vous devez écrire votre propre code R). La pente diffère-t-elle significativement selon les espèces ?
anova(m_iris_spec)Analysis of Variance Table
Response: Petal.Length
Df Sum Sq Mean Sq F value Pr(>F)
Petal.Width 1 430.48 430.48 3294.5561 < 2.2e-16 ***
Species 2 13.01 6.51 49.7891 < 2.2e-16 ***
Petal.Width:Species 2 2.02 1.01 7.7213 0.0006525 ***
Residuals 144 18.82 0.13
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Étant donné que l’interaction entre la largeur des pétales et l’espèce est significative (la p-value de Petal.Width:Species est de 0,0006525), la pente varie d’une espèce à l’autre de manière significative.
2 points en plus
Vérifiez les hypothèses du modèle avec check_model() (vous devez écrire votre propre code R). Pensez-vous qu’une des trois hypothèses principales est violée ? Expliquez pourquoi.
check_model(m_iris_spec)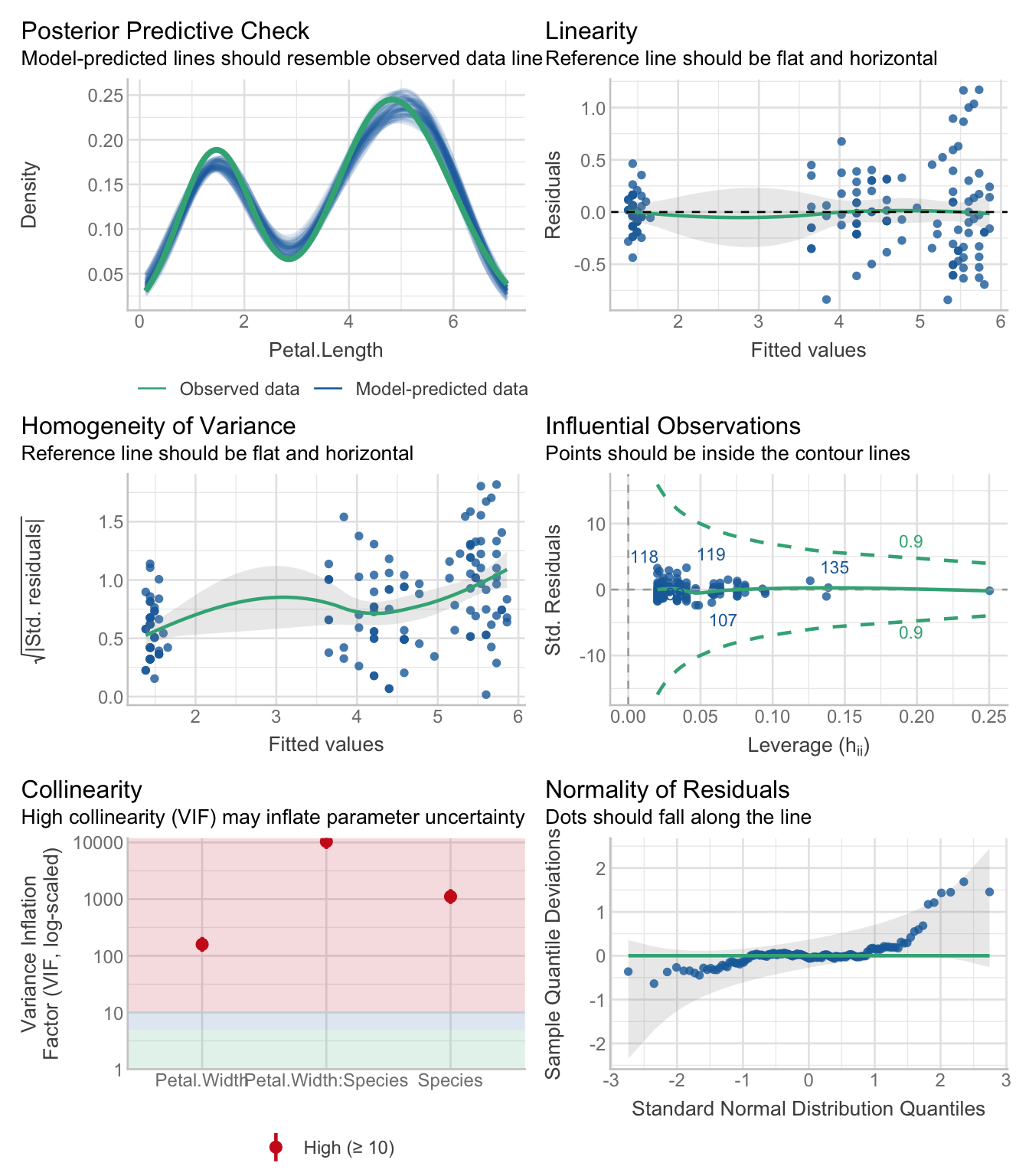
Ces visualisations mettent en évidence certains problèmes dans le modèle. L’hypothèse de normalité est sur le point d’être violée, car on observe un écart important entre les résidus observés et la droite correspondant à une distribution normale. Cependant, l’intervalle de confiance englobe toujours cette droite, ce qui rend cette violation acceptable.
La variance augmente toutefois avec les valeurs moyennes. L’hypothèse d’homoscédasticité semble également violée.
Par ailleurs, vous pouvez analyser un tel ensemble de données de manière plus appropriée en utilisant un modèle GLM avec une distribution gamma (les données ne peuvent être que positives).